1. Sastry, L., Johnson, T., Hobson, M. J., Smucker, B., & Cornetta, K.
Titering lentiviral vectors: comparison of DNA, RNA and marker expression methods. Gene Therapy 2002; 9, 1155—1162.
2. Forghani, B., Hurst, J. W., & Shell, G. R.
Detection of the human immunodeficiency virus genome with a biotinylated DNA probe generated by polymerase chain reaction. Molecular and Cellular Probes 1991; 5, 221—228.
3. Scherr, M., Battmer, K., Blomer, U., Ganser, A., & Grez, M.
Quantitative determination of lentiviral vector particle numbers by realtime PCR. BioTechniques 2001; 31, 520—524.
4. Chengfeng Lei, Jian Yang, Jia Hu, Xiulian Sun
On the Calculation of TCID50 for Quantitation of Virus Infectivity; Virol Sin. 2021 Feb; 36(1): 141–144.
5. LaBarre, D.D., & Lowy, R.J.
Improvements in methods for calculating virus titer estimates from TCID50 and plaque assays. J Virol Methods. 2001 Aug;96(2):107-26. doi: 10.1016/ s0166-0934(01)00316-0.
6. Bramhachari, P.V., Sheela, G.M., Prathyusha, A. M. V. N., Madhavi, M., K. Satish Kumar, K.S., Rao Reddy, N.N., and Chanda Parulekar Berde, C.P.
(2020). Advanced Immunotechnological Methods for Detection and Diagnosis of Viral Infections: Current Applications and Future Challenges. In: Bramhachari, P. (eds) Dynamics of Immune Activation in Viral Diseases. Springer, Singapore. https://doi.org/10.1007/978-981-15-1045-8_17
7. d’Hérelle F.
The bacteriophage and its behavior: Baltimore, Md.: The Williams & Wilkins Company; 1926.
8. Dulbecco R.
Production of Plaques in Monolayer Tissue Cultures by Single Particles of an Animal Virus. Proc Natl Acad Sci USA. 1952; 38(8):747–52.
9. Artur Yakimovich, Vardan Andriasyan, Robert Witte, I-Hsuan Wang, Vibhu Prasad, Maarit Suomalainen, Urs F. Greber;
Plaque2.0—A High-Throughput Analysis Framework to Score Virus-Cell Transmission and Clonal Cell Expansion; PLoS ONE 2015; 10(9): e0138760. doi:10.1371/journal.pone.0138760
10. Ramani et al.
SARS-CoV-2 targets neurons of 3D human brain organoids; The EMBO Journal (2020)39:e106230 https://doi.org/10.15252/embj.2020106230
11. Corman, V. M. et al.
Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Euro Surveill. 2020, Jan.23; 25(3): 2000045; doi: 10.2807/1560-7917.ES.2020.25.3.2000045
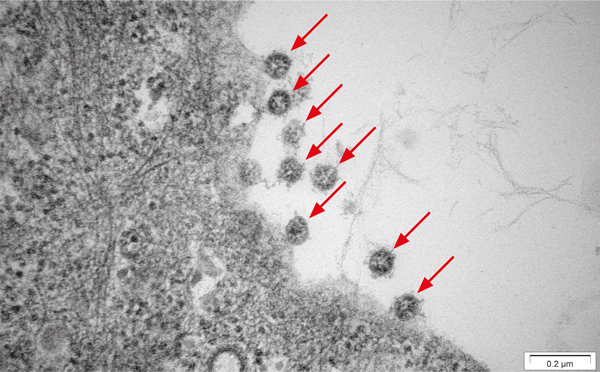
12. Laue M., Kauter, A., Hoffmann, T., Möller, L., Michel, J., Nitsche, A.
Morphometry of SARS-CoV and SARS-CoV-2 particles in ultrathin plastic sections of infected Vero cell cultures 2021. Nature Scientific Reports 11; 3515; https://doi.org/10.1038/s41598-021-82852-7